 Documentation
Documentation
Documetation on the usage of the Protein Structure Annotation Tool (ProSAT+).
What is ProSAT+?
Introduction
ProSAT+ is a tool to explore the relation between sequence and structural properties. It can be used to map and visualize sequence annotations onto a structure using an internet browser like Firefox, Google Chrome, Safari or Microsoft Explorer. For the visualization of the structure, no Java plugin is required. The structure is displayed using JSmol, a JavaScript implementation of Jmol. (Java plugins have recently become very restrictive, due to many security concerns.)
ProSAT+ allows the visualization of a 3D protein structure with highlighted functional regions together with annotations of the functional effects of point mutations. Sequence annotations from UniProt are displayed, but user annotations, provided in a URL to the viewer can also be visualized. Using this approach, one can publish structure sequence mapping annotations using an URL.
The SIFTS resource is used, which provides a residue mapping between PDB and UniProt entries in XML format. Additionally, a BLAST search can be done to add further sequence annotations from evolutionarily related proteins. Further sequence based information can be added, e.g. domain annotations, conservations, and post-translational modifications. The combination of all these information types and the possibility to highlight them simultaneously on the 3D structure helps to analyze protein function and the impact of variations, e.g. mutations, on functional properties.
Changes
- 1.0 Initial version
- 1.1
- Added feature to show molecular surface of protein
- Checkbox on protein name to highlight respective protein chain
Technical information
ProSat+ depends on external services. Monitor service statuses.
Due to the rich set of information displayed in ProSAT+, it is viewed best with a display width of at least 800 px.
ProSAT+ has been tested on the following operating systems:
| Browser/OS | Windows 8, 10, Server 2010 | Linux (Ubuntu 14.04, 15.10) | Mac OS (10.11.4) |
|---|---|---|---|
| Firefox | 45.0 | 38.0,45.0 | 45.0 |
| Chrome | 49.0 | 49.0 | - |
| Safari | - | - | 9.1 |
Elements of ProSAT+
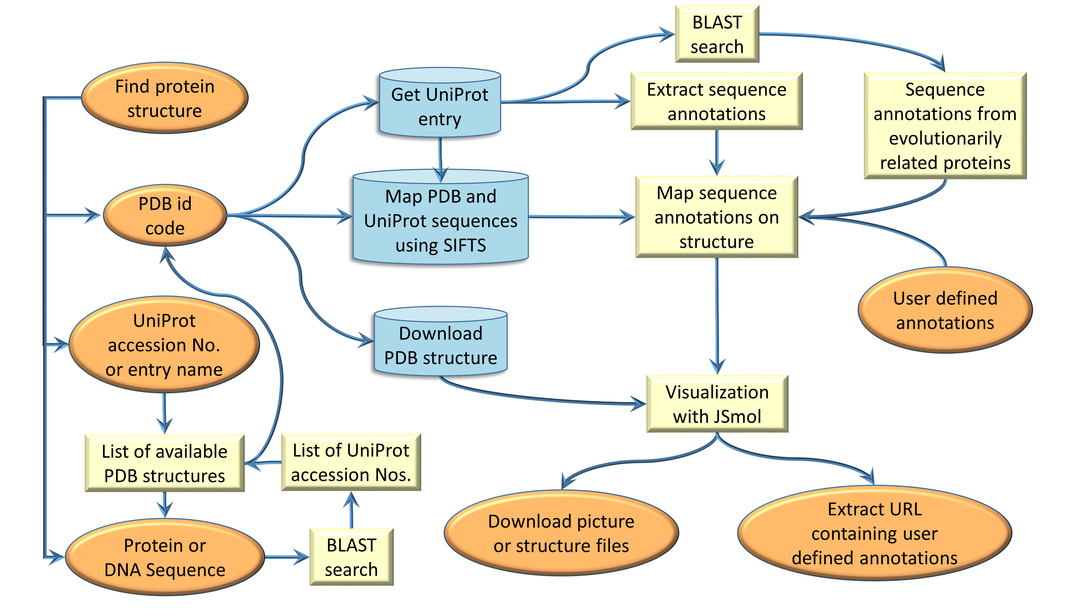
ProSAT+ workflow
The overall workflow and the different database accesses in ProSAT+ are shown in the figure below.
Each step is described in more detail via a tool-tip and some are linked to the corresponding reference web site.
- Orange: User in- and output
- Yellow: Internal processes
- Blue: Database access
Sequence annotation extraction from UniProt
Sequence annotations listed in UniProt for a specific protein are extracted and grouped in different categories. This keyword based grouping method is reused from the previous ProSAT and ProSAT2 versions. The group categories for the respective impact and the corresponding key words are listed in the table below.
| Impact category | List of keywords |
|---|---|
| active site | active site |
| disease | cancer, mild, disease, lethal, severe, patient, leukemia |
| kinetics | turnover, KCat, velocity, vmax, affinity, interaction, Kd, Ki, Km, association, loss of activity, decreased binding, impaired binding |
| binding | binding,zinc finger |
| isoform | isoform, var_seq |
| domain | domain, peptide, region of interest |
| modified residue | modified residue, glycosylation, lipid moiety-binding |
| variant | variant, turnover, KCat, velocity, vmax, affinity, interaction, Kd, Ki, Km, association, mutagensis, mutagen |
| turn | turn |
| helix | helix |
| strand | strand |
| sequence conflict | sequence conflict |
| motif | motif, repeat |
| membrane | transmembrane, intramembrane |
| other | all annotations that could not grouped with the existing keywords |
| user | manually added user defined sequence annotations via URL |
The sequence scroll bar
The horizontal scroll bar of the protein sequence enables a residue specific selection and visualization on the 3D structure.
The scroll bar can be used to scroll through the whole amino acid (AA) sequence of the protein. For each residue, the following
information is given: the residue number according to the UniProt sequence, a checkbox to select and visualize the residue in
the 3D structure, the amino acid and the possible aligned residue (see option 'Find similar sequences'), and all sequence features
available in the UniProt entry.
AA/ is shown in the case of no alignment, otherwise the aligned AA is shown with the colored boxes above the residue representing
the available sequence annotation at that position. Different colours represent different types of sequence features. If the check
box is coloured grey and not white it means that the 3D structure does not contain this amino acid and therefore it can not be
selected and visualized. The background colour of each residue represents the polarity, see
applied colour scheme.
The JSmol visualization
The protein structure is visualized with JSmol, a JavaScript implementation of Jmol, which does not need a Java plugin. This enables a more independant usage as Java plugins have recently become very restrictive, due to many security concerns. JSmol allows to visualize the structure, the ligand, or single residues in various kinds of representation (e.g. cartoon, wire, or spacefilling). Some visualization options are predefined in the 'Configure JSmol' tab at the top of the page. With these options, the visualisation can be changed, pictures of the structure can be saved and downloaded and, for more advanced users, the JSmol console can be displayed. In the JSmol console, it is possible to enter any JSmol command, listed here , which enables a more specific visualization if necessary. The 3D structure can be rotated while keeping the left mouse button pressed. To zoom in and out, the mouse wheel can be used and press Shift + double-click and drag to translate the whole structure. More mouse commands are listed here. A right click on the JSmol window shows further visualization options.
The sequence feature panel
The sequence feature panel is the main tool for visualizing annotations for the given protein. The panel can be opened by clicking the
"Show feature panel" button below the sequence scroll bar, or by clicking on the ProSAT+ button on the top left of the page. In the panel,
a list of different types of sequence features is shown for each chain. For example, all binding site annotated residues can be selected
at once by selecting the box on the 'binding site' level. For single residue annotations, the drop down can be opened by clicking the
arrow on the right. For sequence variants, a link ("_?_") to a more detailed description is given.
The sequence feature panel can be closed again, by clicking on the "Close features panel" at the footer of the page.
Motif search
An input field for a motif search is shown below the sequence panels for the different chains. Regular expressions can be used using this syntax for patterns. When using the Safari browser, searching for the pattern "N[^P][ST][^P]" requires entering "[N][^P][ST][^P]".
If there is a pattern match, the list of matches is given below the input field together with a checkbox. Selecting the checkbox will highlight the corresponding region on the 3D structure as well as marking the corresponding checkboxes in the sequence panel.
Since the motif is searched in the uniprot sequence, it can happen, that matches are not resolved in the 3D structure. In this case, the corresponding checkboxes in the sequence panel will be in disabled state.
User defined sequence annotations via URL
ProSAT+ allows links to structures or UniProt sequences to search for structures. The following URL can be used to search for structures for a given UniProt entry or to link a PDB structure in ProSAT+ directly:
Link to UniProt entry:https://prosat.h-its.org/prosat/prosatuniprot?uniprotacc=P35557
In addition, ProSAT+ offers the opportunity to add user-specific sequence annotations via the URL. This enables the user to define features
for specific residues and, via the URL, it is possible to send these features to other people who can immediately see the defined features
in the context of all the other features on the 3D structure, without having to download the structure or face issues with the residue
numbering. This option can also be useful for publications where one or more residues are of interest. By entering the specific URL in their
web browser, the reader can immediately view the specified sequence features.
In the visualisation mode it is possible to click on the 'Generate URL' menu button at the top of the page. A window will slide in with the
table of all user defined sequence annotations. The user can add single annotations by using the input fields or by pasting many annoations
at once in the following format:
UniProt accession number:residue number(s) separated by comma:name:Description
Instead of the colon a tab separation is also possible. One feature can include more than one residue (comma separated and ranges are defined
with a dash (e.g. 1-15))and, if more than one sequence feature is added, a "|" or a newline is necessary as a separator.
All annotations are listed in the editable table and the final URL can be automatically generated.
When using the specific URL the user specified features (mentioned as 'user') are visualized and listed in the sequence scroll bar and
the feature panel.
Using URL shortner like https://goo.gl/ allows to use the long URLS in texts like publications.
It is also possible to generate a URL containing user-specific sequence annotations without a UniProt accession number. In this case the given residue number(s) refer to the sequence numbers in the PDB file. Instead of a UniProt accession number the keyword 'PDB' is entered and seperated by a point the PDB chain id is given. Such URL could look like this https://prosat.h-its.org/prosat/prosatexe?pdbcode=1hti&user=PDB.A:102:name:ThisisOneFeatureDescription where residue number 102 is in this case the Phe140 in the UniProt sequence.
Known problematic cases
We tried to implement ProSAT+ to be as robust as possible for all PDB structures, sequences, and mapping cases. Nevertheless,
the following special cases currently lead to some problems. We are working on solutions for them. If you find other cases that cause
problems or see any errors etc., we would be happy to receive your report by email to:
mcmsoft(at)h-its.org
- Sometimes there is no UniProt entry for a given PDB structure or for parts of the PDB structure. This can, for example, be the case for DNA or a synthetic construct. The PDB entries 4ymn and 1uw1 are examples. In such cases, the user is informed that no mapping exists and the sequence scroll bar contains the sequence from the PDB file.
- Sometimes multiple UniProt entries are assigned to one chain in the PDB structure (eg. fusion proteins). At the moment, ProSAT+ is not able to handle such cases and no sequence mapping data is available and consequently no sequence annotations. Examples are the PDB entries 4z4m and 4zj8.
- In case there is no UniProt entry for a given PDB structure we use the sequence in the PDB file. In case such PDB file also contains an amino acid which is assigned with a residue number containing an additional character (e.g. residue 52A in PDB id 1yqv) the numbering in the sequence scroll bar is not the same in the PDB file anymore, but the shift can be seen in the label of the residues.
ProSAT+ example usage
Example 1: Hemoglobin
This example will show and explain most of the features that the ProSAT+ web server offers.
At the beginning of a project, the identity of the target protein is often known. Therefore, it is useful to check what is already known about the protein at the structure and sequence level. ProSAT+ helps to do exactly this and goes beyond by including sequence annotations from evolutionarily related proteins. If there is no 3D structure available for the protein of interest, ProSAT+ helps to find an appropriate structure.
Our example protein is hemoglobin and if we know that the PDB id 1j7y contains the 3D structure, we can type the PDB id into the text field and click on 'Check PDB'.
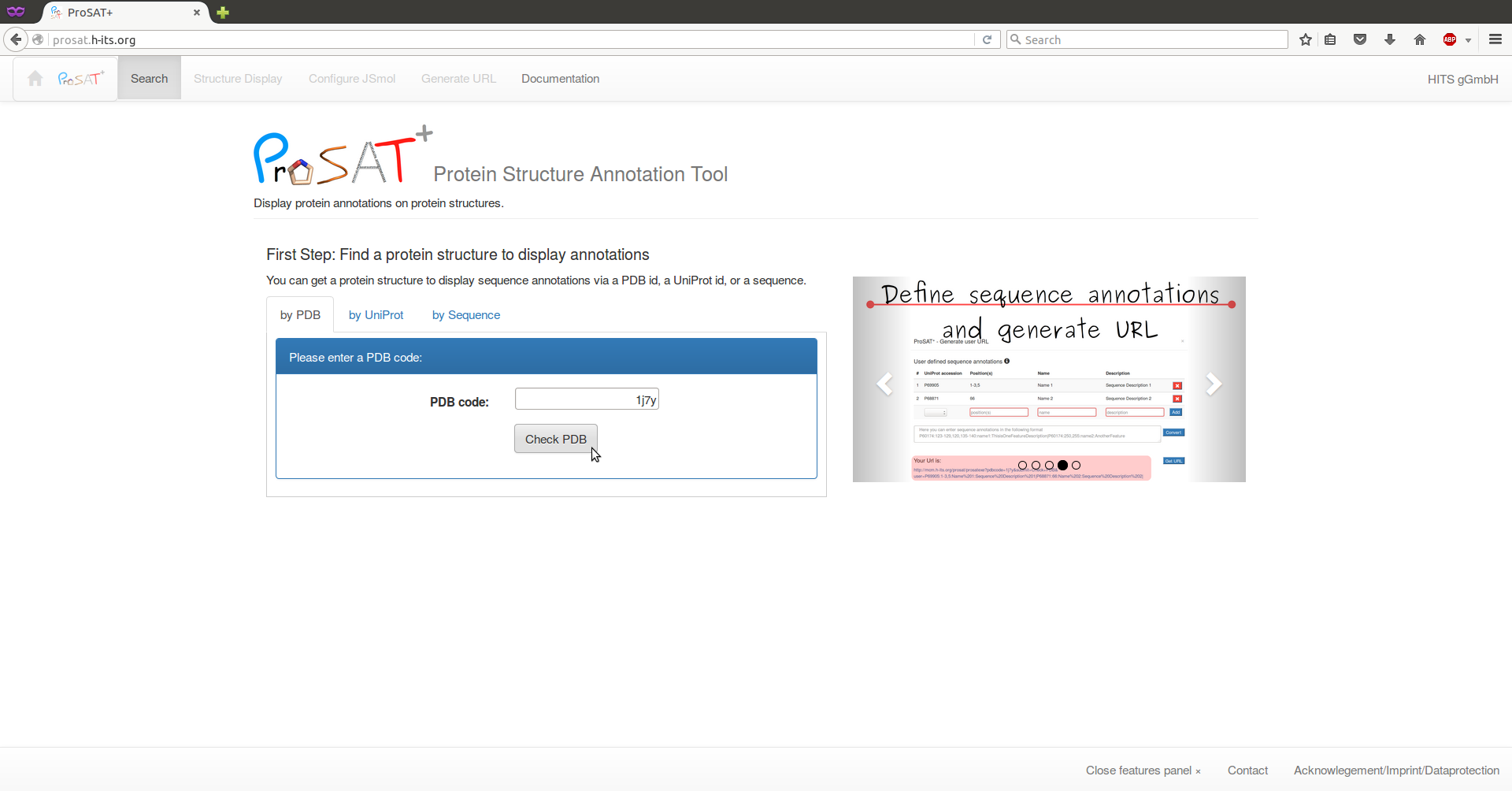
If we only know the UniProt Id, we can search for all available 3D structures via the UniProt accession P69905 and select e.g. 1j7y from the list. In this case, we click on the tab 'by UniProt' and search for the id. A list of all PDB structures containing some structural information is shown and one PDB id has to be selected. If no structure is available for a target protein, a BLAST search will be done and a list of evolutionarily related proteins with 3D structures available will be shown from which structures can be selected.
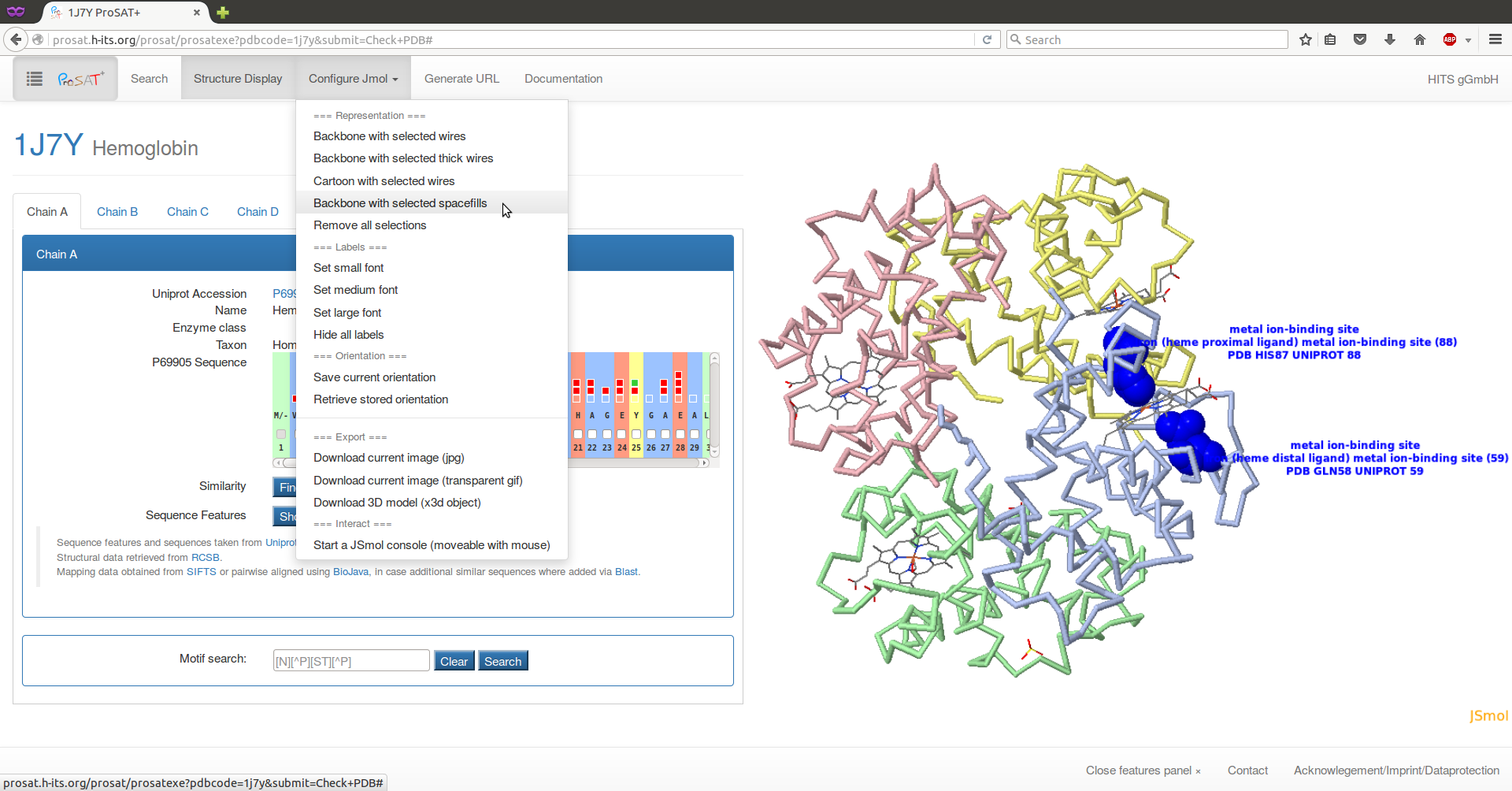
Here, we use the PDB id 1j7y. After selecting 'check PDB', the structure is shown on the right and information about the respective chains is shown on the left. Select the second residue ("V") in the sequence scroll bar, to visualize the residue in the 3D structure together with the SIFTS mapping information about the numbering in the respective sequences. Here, one advantage of ProSAT+ is visible: the residue numbering in the PDB structure is different from the numbering in the UniProt entry, but the web server automatically maps all sequence annotations on the correct residue in the 3D structure.
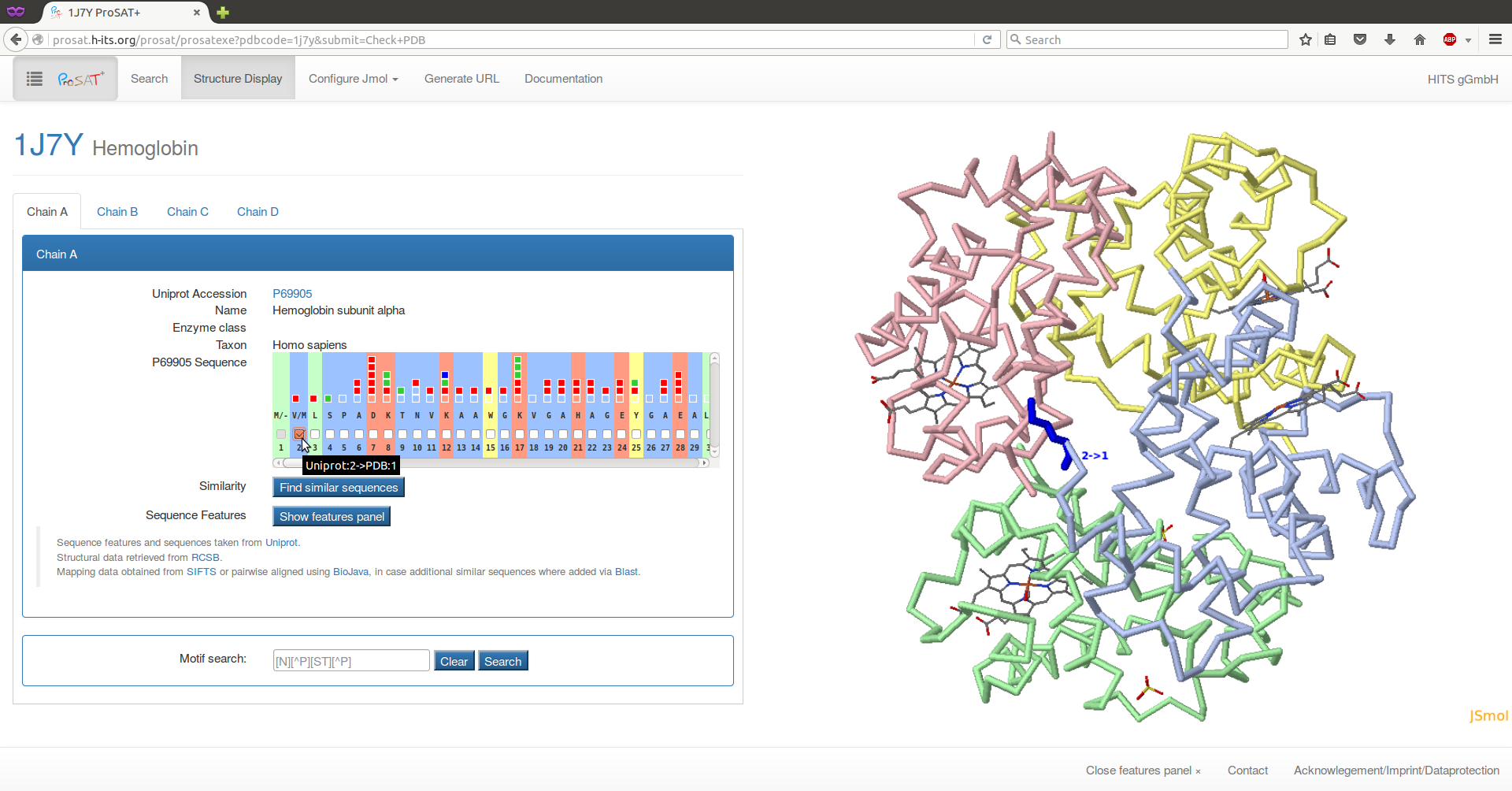
Hemoglobin is a heterotetramer that consists of two alpha (P69905) and two beta (P68871) subunits. Here, the sequence features of both subunit types are collected and can be visualized simultaneously in the 3D structure, enabling an analysis of sequence features in the interface between the two subunits.
By clicking on the 'Show features panel' or the ProSAT+ button on the top left of the page, the feature panel will slide in.
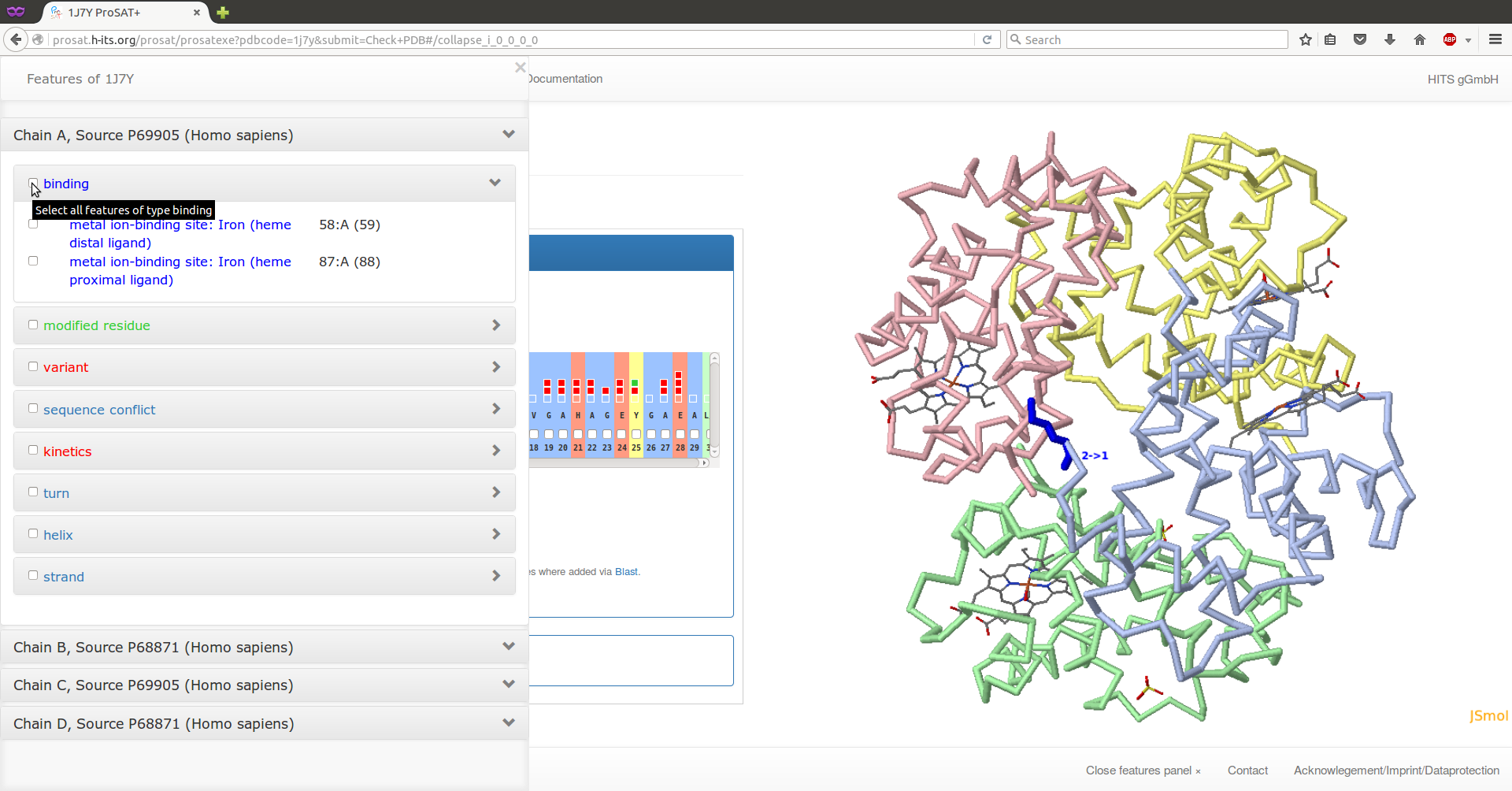
Select the two residues annotated as "binding" on chain C (alpha subunit) by using the drop down lists (click on the arrows on the right). Now the representation of the protein should be changed. Click on the 'Configure JSmol' menu button at the top of the page and select a different representation, e.g. 'Backbone representation with selected spacefills'. The two binding site residues are now displayed as space filling, the protein as backbones, and the ligand as lines.